US FDA 21 CFR 820.30 Design Control Requirements
Complying with US FDA 21 CFR 820.30 Design Controls is essential for medical device manufacturers planning to enter the U.S. market. This regulation, part of the FDA’s Quality System Regulation (QSR), sets detailed requirements for the design and development of Class II and Class III medical devices. While expertise in FDA QSR is often U.S.-based, global consulting firms like Operon Strategist offer specialized support to manufacturers worldwide. We assist in implementing FDA-compliant design control processes, preparing comprehensive technical documentation, and aligning development practices with QSR standards ensuring a smoother path toward FDA 510(k) or PMA submissions.
What is US FDA 21 CFR 820.30 Design Control?
FDA 21 CFR 820.30 outlines the design control requirements under the U.S. FDA Quality System Regulation (QSR) for medical devices. It ensures that the design process consistently results in safe, effective, and regulatory-compliant products.
Manufacturers including those in the Netherlands must establish and maintain a structured design and development plan. This includes design inputs, outputs, verification, validation, and reviews to confirm that the final device meets user needs, performs as intended, and complies with FDA requirements.

Design Control 21 CFR 820.30 Process for Medical Devices :
Build up and maintain a plan that describes the design and development activities and allocates the individual obligations for each activity. Guarantee you review, update and approve the plan until the device design is completed, verified and validated.
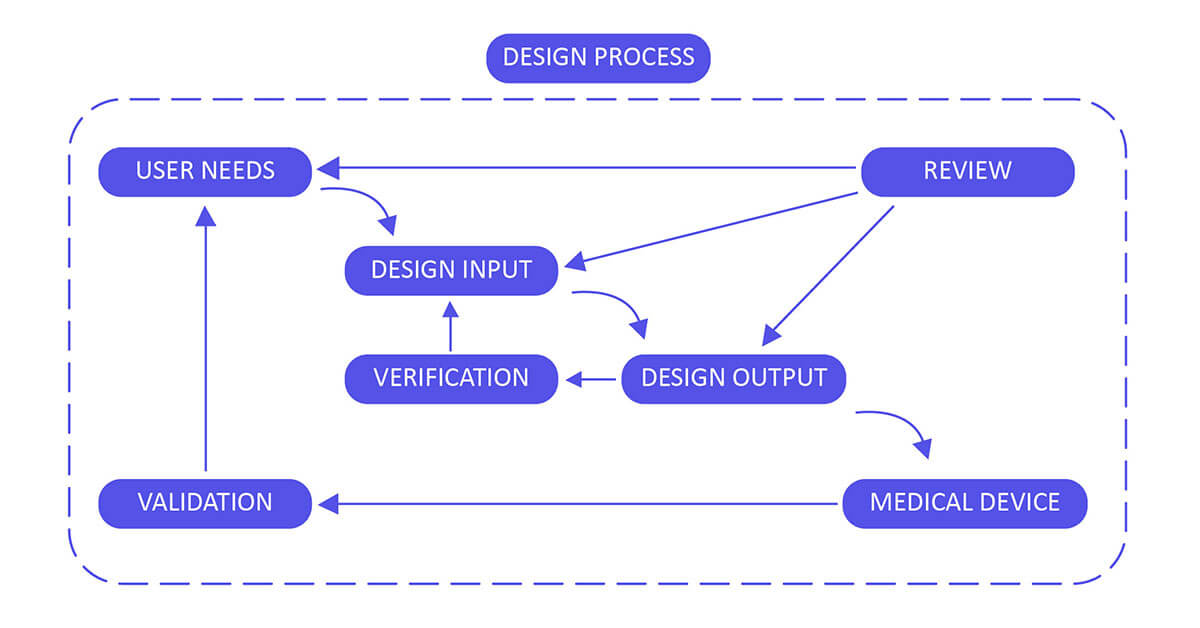
Design Input :
Utilize performance, safety, business economics, outputs of risk management and regulatory requirements as a basis to plan the device with the goal that its motivation and the proposed utilize are clear. The input may also come from surveying your customers( For example, clinicians, nurses, patients).
Design Output :
Design output methods or particulars need to stipulate or refer to the design input document developed by the team and need to identify the critical measures/outputs for the best possible capacity of the device. These incorporate the tests and strategies that may have been produced, adjusted or used to show conformance with the characterized configuration inputs. Examples of design outputs may include:
- The device itself.
- The user manual.
- Specifications A Risk Analysis Study results (For examples, validation and biocompatibility studies, storage).
- Technical Files.
Design Review :
Confirm the design, or identify at an opportune time and right any insufficiencies distinguished at other plan and improvement phases. Two common types of review are hazard analysis, and failure mode and effect analysis.
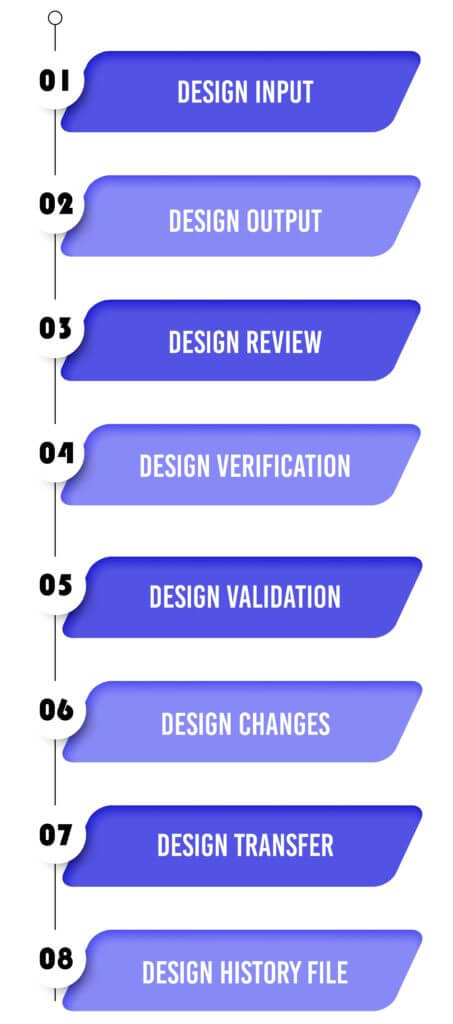
Design Verification :
Confirm the device outline by means of examination and target prove, verify that the design outputs meet the plan inputs. Design verification activities must be arranged and routinely analyzed and the outcomes must be documented.
Design Validation :
Approve the device design plan by means of examination and target prove, affirm that the last outline yield reliably meets the particular planned utilize. Design validation should follow successful design verification. Since outline check is directed while the plan work is being performed, design validation confirms that the medical device meets its intended use. Generally, this is set up through in vitro execution, practical testing, creature testing and additionally in vivo clinical assessments and trials.
Design Changes :
Guarantee that all plan changes are distinguished, documented, approved, verified, reviewed and endorsed before usage.
Design Transfer :
Ensure that the design of the medical device can be correctly translated into production specifications (that is, advancing successfully from product development to manufacturing).
Design History File :
The design history file (DHF) aggregates confirm (that is, the history of the design) that demonstrates that the outline was created as per the outline of design control― specifically, the design and development plan, or the outline change design.
Operon Strategist’s Role in Supporting Medical Device Manufacturers
Operon Strategist offers expert consulting services for implementing FDA 21 CFR 820.30 Design Controls, helping medical device manufacturers align with both US FDA regulations and ISO 13485:2016 requirements.
We support manufacturers in establishing robust design and development processes that meet international compliance standards. Whether you’re aiming for U.S. market entry or looking to strengthen your internal Quality Management System (QMS), our team ensures a structured, risk-based, and regulation-ready design control framework.
Master 21 CFR 820.30 with Proven Regulatory Expertise
FAQs
What is FDA 21 CFR 820.30 Design Control?
It's a regulatory requirement under the U.S. FDA Quality System Regulation (QSR) that mandates a structured design and development process for Class II and III medical devices to ensure safety, effectiveness, and compliance.
Who needs to comply with 21 CFR 820.30?
All manufacturers developing Class II or Class III medical devices intended for the U.S. market must implement design controls as part of their Quality Management System (QMS).
How does ISO 13485 relate to 21 CFR 820.30?
While ISO 13485:2016 provides an international standard for medical device QMS, 21 CFR 820.30 is a U.S.-specific requirement. Both overlap significantly, but 820.30 places more emphasis on documentation and traceability in the design phase.
Why is design control important in medical device development?
Design control ensures that your device is developed systematically, meets user needs, performs safely and effectively, and complies with FDA requirements—reducing risks of product recalls or approval delays.
Is design control required for Class I devices?
Generally, Class I devices are exempt from 21 CFR 820.30, but exceptions exist based on the specific product type. Manufacturers should verify classification and exemptions with regulatory experts.