
US FDA 21 CFR 820.30 Design Control Requirements
Regarding consultants specializing in US FDA 21 CFR 820.30 Design Controls in Saudi Arabia, it’s important to note that expertise in this specific regulation may not be as prevalent outside the United States, especially in regions like Saudi Arabia. However, there are consultancy firms, both within Saudi Arabia and internationally, that offer expertise in quality management systems, regulatory compliance, and medical device development.
US FDA 21 CFR 820.30 Design Control - An Overview
FDA 21 CFR Part 820.30, also referred to as the Design Control Requirements, represents a critical stage in the creation of a medical device. A defective design holds the potential to make the device inefficient or hazardous, resulting in rejection or lack of approval from regulatory bodies. To address this, a well-defined design control process is instigated during the design phase and seamlessly integrated into the Quality System Requirement. These design controls encompass systematic and coherent steps aimed at guaranteeing that the ultimate product adheres to the planned design and fulfills the needs and standards of its users.

According to 820.30 guidance manufacturers of medical devices from Saudi Arabia or from any other country should establish or maintain plans that describe design and development activities.
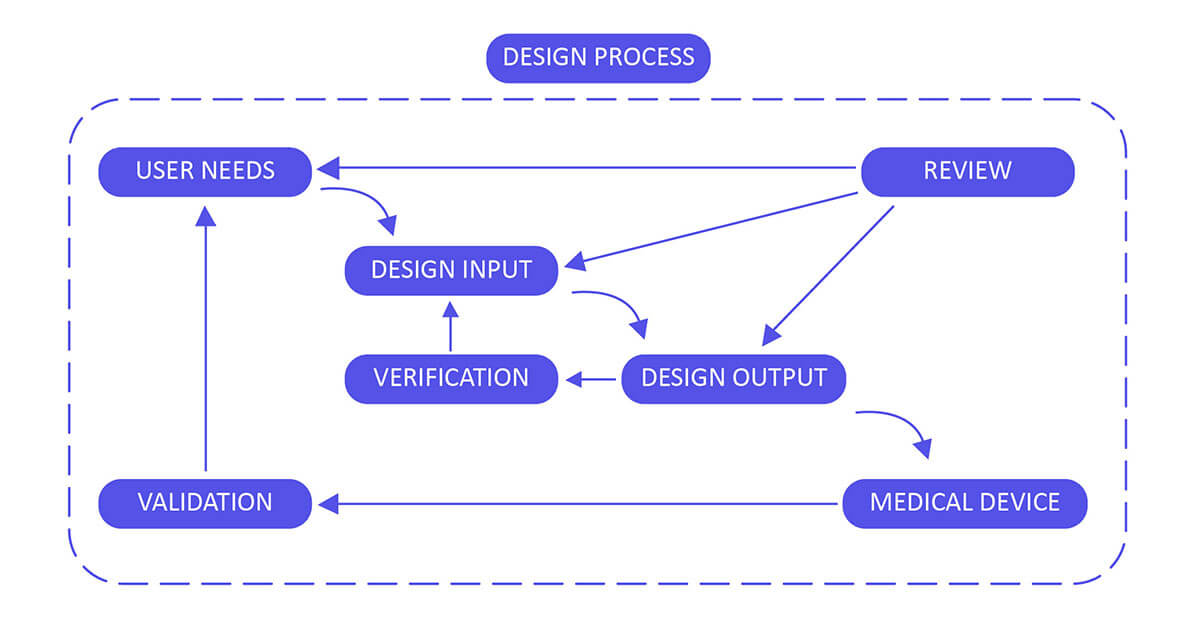
Design Control 21 CFR 820.30 Process for Medical Devices:
Develop and uphold a comprehensive plan outlining all design and development activities, clearly assigning specific responsibilities for each task. Ensure the plan undergoes consistent review, updates, and approval at regular intervals throughout the entire design process. This includes stages from the initial concept to the ultimate finalization, verification, and validation of the device design. Regularly assess and enhance the plan to reflect any changes or advancements, ensuring it aligns with evolving requirements and stays in sync with the overarching project goals.
Looking For Regulatory Consultants in Saudi Arabia?
Design Input :
Establish a robust framework for planning the device, grounded in several key pillars: performance, safety considerations, business economics, outputs derived from risk management, and compliance with regulatory requirements. This foundation will ensure a well-defined purpose and clear proposed use for the device. To enhance this process, solicit valuable input from diverse stakeholders, such as clinicians, nurses, and patients. This collaboration aims to align the design of the device with real-world needs and expectations, leveraging their insights to enhance functionality, usability, and overall effectiveness. Incorporating feedback from these stakeholders allows for a more comprehensive understanding of the device’s intended purpose, facilitating a design that better serves its intended users and the broader healthcare ecosystem.
Design Output :
Design output methods and specifications should clearly refer to the team-developed design input document and identify critical measures and outputs for optimal device performance.
- The device itself.
- The user manual.
- Specifications A Risk Analysis Study results (For example, validation and biocompatibility studies, storage).
- Technical Files.
Design Review:
Validate the design by either confirming it as is or addressing any identified deficiencies early in the design and development process. Two typical forms of review include hazard analysis and failure mode and effect analysis (FMEA). These reviews help ensure the design’s safety and effectiveness.
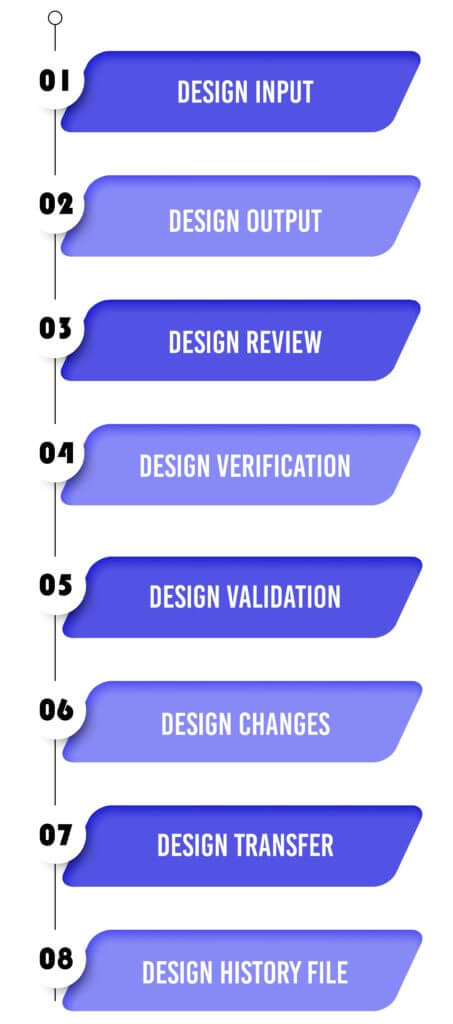
Design Verification :
Validate the device design through examination and objective evidence, ensuring that the design outputs align with the initial design inputs. Plan and conduct design verification activities in a systematic manner, with regular reviews, and comprehensively document the outcomes of these activities.
Design Validation :
Validate the device design plan through examination and objective evidence to ensure that the final design output consistently aligns with the intended use. Design validation should be conducted following successful design verification. While design verification occurs during the design process, design validation verifies that the medical device indeed fulfills its intended purpose. This is typically achieved through in vitro performance, practical testing, animal studies, and in vivo clinical assessments and trials.
Design Changes :
Ensure that all design changes are identified, documented, approved, verified, reviewed, and authorized before being implemented. This process helps maintain control over design modifications and ensures that they meet the necessary requirements and standards.
Design Transfer :
Ensure that the medical device’s design can be accurately translated into production specifications, allowing for a seamless transition from product development to manufacturing. This alignment between design and production is essential for the efficient and effective manufacturing of the device.
Design History File:
The design history file (DHF) aggregates confirm (that is, the history of the design) that demonstrates that the outline was created as per the outline of design control― specifically, the design and development plan, or the outline change design.
Operon Strategist Roll
Operon Strategist the leading medical device regulatory consultant providing consultation for 21 CFR 820.30 design control with extensive experience and the practical implementation of design controls regulation for developing new design control processes or for making improvements to existing processes. We assist medical device manufacturers of Saudi Arabia in design controls as per FDA and ISO 13485:2016 that can be mapped to the process that works best for the organization and the product being developed. If you need any help in setting up a design control system or wish to modify an existing system in order to align with ISO 13485 or FDA design controls, feel free to contact us.