
US FDA 510(k) Consultant for Medical Devices and IVDs
What is US FDA 510(k) for Medical Devices?
The FDA 510(k) is a premarket submission process required for certain medical devices to demonstrate they are substantially equivalent to a legally marketed predicate device. It applies primarily to Class II and some Class I devices and includes details about the device’s design, intended use, and performance testing. A 510(k) submission is necessary for new devices or significant modifications to existing ones, unless exempt. If cleared, the device can be marketed in the U.S., though 510(k) clearance indicates equivalence to a predicate device, not FDA “approval” or guaranteed performance. The process typically takes about 90 days.
Types of FDA 510(k)
- Traditional FDA 510(k) – Also known as original 510(k) and used in any circumstances.
- Abbreviated FDA 510(k) – When the submission relies on FDA guidance documents, demonstration of compliance with special controls for device type, voluntary consensus standard.
- Special FDA 510(k) – When there is a modification in the device however the modification should not affect the intended use.
Looking for US FDA Registration for Medical Devices and IVDs?
Fill the Form or Mail Us to: enquiry@operonstrategist.com
US FDA 510(k) Clearance, Submission & Premarket Approval Consultant
Operon Strategist is a leading FDA 510(k) Consultant, providing FDA 510(k) Clearance process consulting to clients in India and other countries to register SBU (Small Business Unit), as applicable. We also assist with the establishment registration and device listings for products that are low risk and do not require special controls under the US FDA. New medical devices for registration with the US FDA require Premarket approval or De Novo approval.

Understanding US FDA Registration for Medical Devices and IVDs
In the United States, the Food and Drug Administration (FDA) regulates medical devices and in vitro diagnostic (IVD) products to ensure their safety and effectiveness. This oversight is critical to protect public health and ensure that products entering the market meet rigorous standards. Here’s a comprehensive overview of the FDA registration process for medical devices and IVDs:
Medical Device FDA 510(k) Approval Overview
FDA 510(k) approval for medical devices is a regulatory process through which the U.S. Food and Drug Administration (FDA) evaluates and authorizes the marketing of medical devices in the United States. The approval process aims to ensure the safety and effectiveness of medical devices before they reach the market. It involves a thorough review of the device’s design, performance, labeling, and manufacturing processes. There are different pathways for approval, including 510(k) clearance, Premarket Approval (PMA), and the De Novo classification process, depending on the device’s level of risk. Compliance with US FDA registration is essential for companies seeking to market and distribute medical devices in the U.S.
USFDA 510(k) Clearance Process for Medical Device
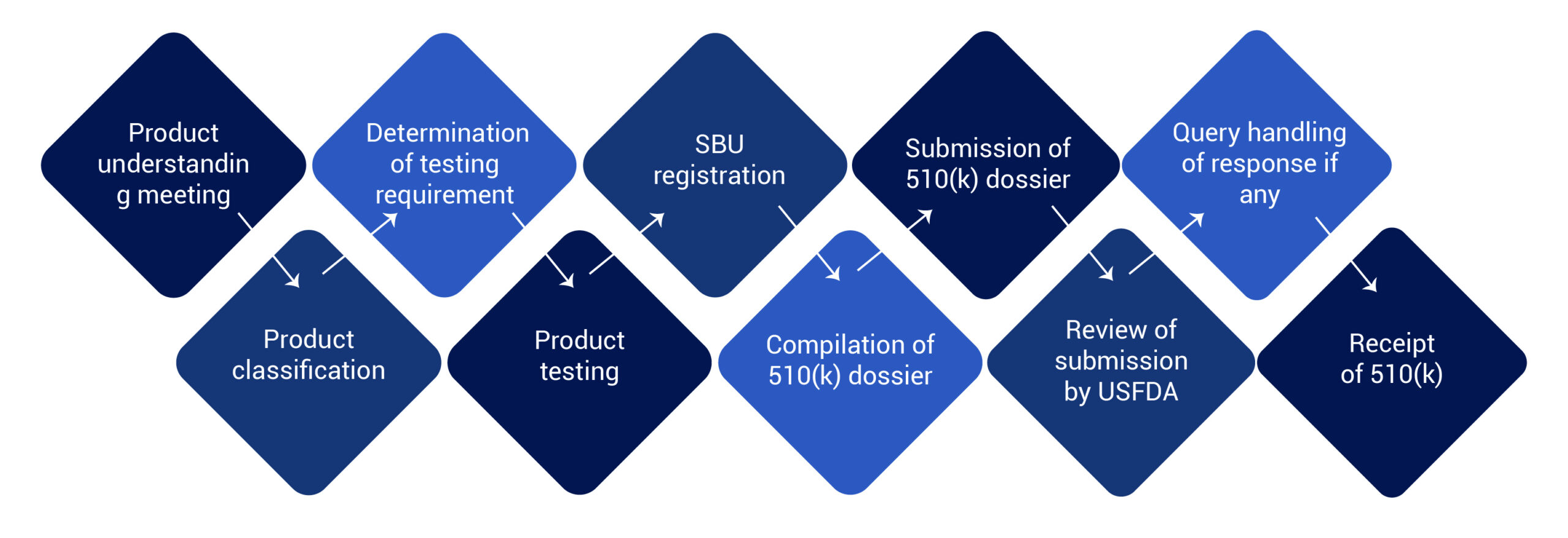
The FDA 510(k) process involves medical devices study and its comparison to a similar product already selling in the US market. This similar product is also known as a predicate device. 510(k) file or dossier must be prepared and submitted to the US FDA for their review.
Post submission the FDA clears the medical devices and issues a 510(k) number. This number with product summary and details are uploaded to the 510(k) database. The database is accessible to the public on www.fda.gov
Looking for FDA 510(k) Consultant Near You?
Device Identifications Points for FDA 510(k)
A device is considered identical only if, in comparison to a predicate it
- The device has to have the same intended use as the predicate; and
- Also, the device should have the same technological characteristics as the predicate;
- Deliberate use should remain the same as the predicate
- Whether the device has different technological characteristics and does not raise different questions of safety and effectiveness
- The data submitted to the FDA reveals that the device is at least as safe and effective as the legally marketed device.
Read More: Tips to Locate a Predicate Device for 510(k)
Who Needs FDA 510(k) and Why?
FDA 510(k) is required to determine whether the device is safe and effective and hence permissible to legally sell in the US market. FDA 510(k) Guidance gives an idea of whether the product is safe and as per the safety rules of the FDA or not.
- 510(k) is required for Class I(Non Exempt) and Class II medical devices.
- Domestic medical device manufacturers trying to introduce new devices to the US market
- Re-packers or Re-labeler’s who make labeling changes
How Can Operon Strategist Help in US FDA Registration and in FDA 510(k) Process?
We have a professional team of experts who closely interact with notified bodies and are completely aware of the regulatory updates necessary for the device to comply.
As an FDA 510(k) consultant, our team will assist you in:
- Interpret FDA regulations and Ensure compliance with FDA standards and guidelines.
- Assist in compiling the necessary documentation for 510(k) submission.
- Identifying predicate device
- Evaluating the substantial equivalence
- Creation of 510(k) technical documents file
- Act as a communication link between clients and the FDA, responding to FDA queries and requests for information.
- Our QMS specialized team helps manufacturers comply with 21 CFR part 820 QSR requirements.
To discuss your support needs, you can contact us at enquiry@operonstrategist.com or you can WhatsApp us at 8767980322 your queries we answer them shortly.
FAQs
What is US FDA 510(k)?
The USFDA 510(k) process is a premarket submission that demonstrates the safety and effectiveness of medical devices. It is necessary to demonstrate that the product is substantially equivalent to one that is sold legally in the US..
How long does it take to obtain 510(k) clearance?
Generally, the FDA aims to review and process a 510(k) submission within 90 days. But the time required to obtain 510(k) clearance can vary depending on various factors, including the complexity of the device, the completeness of the submission, and the workload at the FDA.
How do I know if a medical device is FDA approved?
To determine if a medical device has received FDA clearance, you can search for the device in the FDA's public database called the 510(k) Premarket Notification Database. This database allows you to look up specific devices and view their clearance status and relevant information. Additionally, you can consult the device manufacturer or contact the FDA directly for confirmation of a device's clearance status.