Medical Device Design Documentation in Germany – Operon Strategist
What is Medical Device Design Documentation?
Medical Device Design Documentation is a critical requirement for manufacturers seeking compliance with EU MDR and ISO 13485 standards. It involves structured records that demonstrate how a medical device was designed, developed, verified, and validated to ensure safety and performance.
At Operon Strategist, we help manufacturers in Germany prepare comprehensive Design and Development Documentation that supports CE Mark approvals and audit readiness. Our experts ensure that your technical files, design history files, and risk management documentation are complete, compliant, and aligned with regulatory requirements.
Whether you are launching a new medical device or updating existing product documentation, Operon Strategist ensures that every stage of your design process is well-documented and audit-ready, reducing approval delays and enhancing market access.
Let's Grow Your Business Together
Why Medical Device Design Documentation Matters?
A robust documentation system ensures:
EU MDR & ISO 13485 compliance for German manufacturers.
Traceability of design decisions and design control activities.
Risk reduction by identifying flaws before market launch.
Efficient approvals with audit-ready documentation packages.
Market acceptance by proving safety, functionality, and usability.
Without comprehensive design documentation, companies risk delays, non-compliance, and financial setbacks.

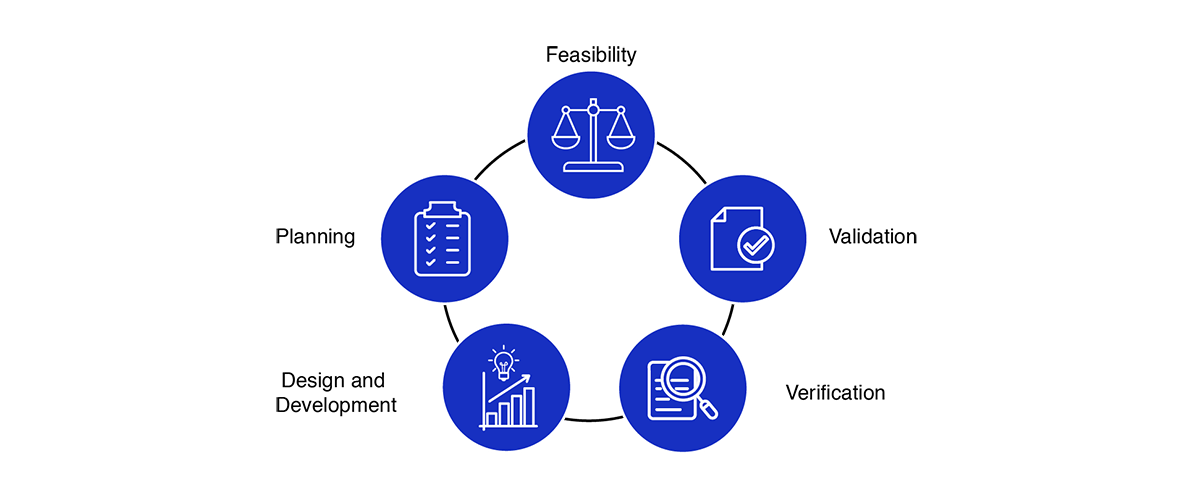
The Medical Device Design and Development Process
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
We support manufacturers in Germany with a step-by-step documentation process that aligns with EU MDR, FDA, and ISO standards.
Core phases include:
Feasibility Study – Assess market needs, risks, and technical possibilities.
Planning – Define design strategy, responsibilities, and compliance path.
Design & Development – Document design inputs, outputs, and prototypes.
Verification – Confirm that the design meets specifications.
Validation – Ensure safe performance in real-world conditions.
Design Transfer – Move seamlessly from design to production.
Post-Market Surveillance – Maintain records for feedback and improvements.
Each stage generates required Medical Device Design Documentation that regulatory authorities expect during audits and submissions.
Design Control and Documentation
Design control is at the heart of compliance. In Germany, under EU MDR 2017/745, manufacturers must prove that devices are safe, effective, and backed by structured documentation.
Key aspects include:
Capturing design inputs and outputs.
Building a Design History File (DHF).
Maintaining risk management documentation (ISO 14971).
Recording verification and validation activities.
Updating documents with every design change.
By implementing proper design control, manufacturers can ensure their devices meet customer needs and regulatory expectations.
Medical Device Design Services & Product Development Process?
- Feasibility
- Planning
- Design and development
- Verification
- Validation
Medical device design services and development process encourages an early focus on clear problem definition and de-risking a wide variety of potential solutions. By later phases, the funnel of medical device design options narrows significantly, converging on a final product that has been thoroughly shown to meet the customer needs and is ready for distribution.
Standards for Medical Device Design Documentation
To achieve compliance in Germany and beyond, documentation must align with global standards:
EU MDR 2017/745 – European medical device regulation.
ISO 13485 – Quality management for medical devices.
ISO 14971 – Risk management requirements.
IEC 62304 – Medical device software lifecycle documentation.
FDA 21 CFR Part 820.30 – U.S. design control regulation.
Meeting these standards ensures approval from Notified Bodies in Germany and other global regulators.
The Medical Device Design and Development Services Includes :
Combination Product - Drug - Device
Each manufacturer of Drug Device combination products (e.g. Drug, device combination products like prefilled syringes, applicators of the tropical products) shall have adequate design and development activity done so as to prove the adequacy of the safety and efficacy of the product. The medical device design and development activity is the systematic methodology, which establishes the proper medical device design and development.
Medical Device Design Control
After conceptualizing a new medical device, the next step in its product advancement is the design. This is the most important stage in the advancement of a medical device since a defective plan may prompt it being inadequate or dangerous (that is, not affirmed or cleared by the administrative organization). At the medical device design stage, an outline control process should be started and actualized as a feature of the quality system requirement.
Our Medical Device Documentation Services in Germany
At Operon Strategist, we help medical device companies in Germany develop and maintain compliant documentation systems.
Our services include:
Full Medical Device Design Documentation packages.
Design And Development Documentation for regulatory submissions.
Audit-ready records for Notified Bodies and FDA inspections.
Risk management documentation aligned with ISO 14971.
Support for CE Marking, FDA clearance, and ISO 13485 certification.
We act as your consulting partner to ensure timely, cost-effective, and error-free compliance.
Why Choose Operon Strategist in Germany?
10+ years of regulatory consulting expertise.
Deep knowledge of EU MDR compliance and German market requirements.
Proven success with ISO 13485 and CE Mark documentation.
End-to-end support for startups and established manufacturers.
Commitment to faster approvals and sustainable compliance.
Don't Miss Any Opportunity! Regulate Your Device With Ease
FAQ'S
Medical Device Design Documentation is the structured set of records that demonstrate how a medical device was designed, developed, verified, and validated. It ensures compliance with EU MDR and ISO 13485 requirements for safety and performance.
CE Marking requires complete design and development documentation to prove regulatory compliance. Proper documentation demonstrates that the device meets safety, quality, and performance standards, which is essential for market approval in the EU.
The documentation typically includes the Design History File (DHF), Design and Development Plan, Risk Management File, Technical File, and records of design verification and validation. These documents together provide evidence of a compliant design process.
Operon Strategist helps manufacturers in Germany prepare complete, audit-ready design and development documentation aligned with EU MDR and ISO 13485. We ensure that technical files, design history files, and risk management documentation are fully compliant and ready for CE Mark submission.