US FDA 510(k) Consultant for Medical Devices and IVDs
US FDA 510(k) Consulting Services for Omani Medical Device Manufacturers
If you are a medical device or IVD manufacturer in Oman planning to enter the United States market, obtaining US FDA 510(k) clearance is a critical regulatory requirement. Operon Strategist is a trusted US FDA 510(k) consultant providing end-to-end regulatory consulting and submission support to help manufacturers achieve compliant and timely FDA clearance.
Our expert team ensures that your medical device or IVD meets all US FDA 510(k) regulatory requirements, enabling safe, effective, and legally compliant access to the U.S. medical device market.

What Is the US FDA 510(k) Process?
The US FDA 510(k) process is a premarket submission to the U.S. Food and Drug Administration that demonstrates a medical device is substantially equivalent to a legally marketed predicate device in the United States.
A US FDA 510(k) submission is required for:
- Most Class II medical devices
- Selected Class I (non-exempt) devices
- In Vitro Diagnostic (IVD) devices, including test kits, reagents, and analyzers
Without FDA 510(k) clearance, these devices cannot be legally sold or distributed in the U.S.
Let’s Connect! Your Queries, Our Expertise!
Who Needs a US FDA 510(k) Submission?
A US FDA 510(k) submission is required for manufacturers intending to market regulated devices in the U.S., including:
- Diagnostic, therapeutic, and surgical Class II medical devices
- Certain Class I medical devices requiring clearance
- IVD products, such as assays, reagents, analyzers, and diagnostic kits
For Omani medical device manufacturers, successful US FDA 510(k) clearance is essential for U.S. market access and global regulatory credibility.
US FDA 510(k) Clearance Process:
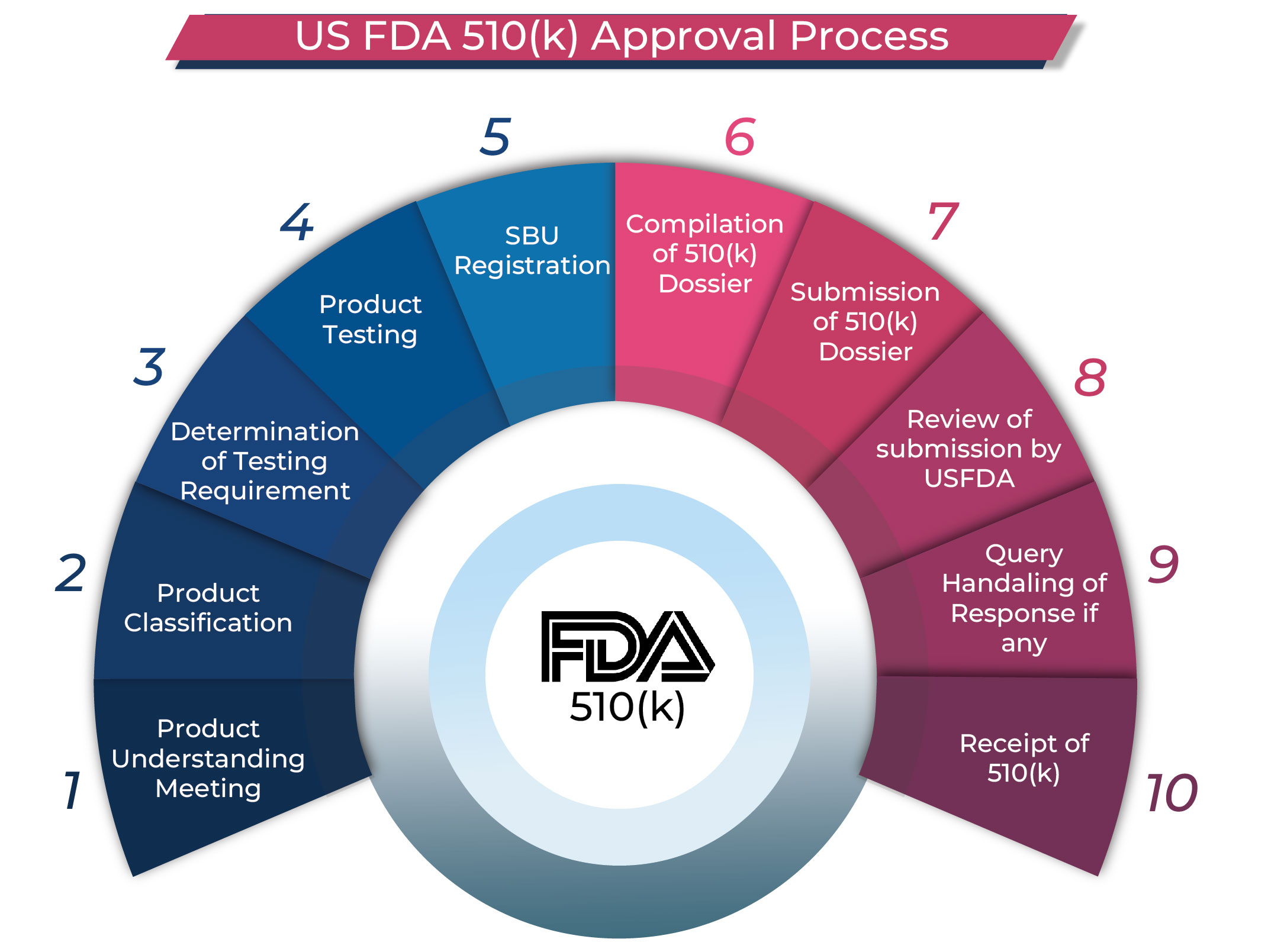
The US FDA 510(k) clearance process involves submitting a detailed dossier to the FDA, comparing the new medical device to a predicate. Once the FDA approves the application, a 510(k) number is issued, and the device is listed on the public FDA database, allowing for legal sale in the U.S.
What Is US FDA 510(k) Clearance?
US FDA 510(k) clearance is a regulatory authorization confirming that your medical device is as safe and effective as a legally marketed predicate device. It applies to many Class I (non-exempt) and Class II medical devices before commercial distribution in the U.S.
This process ensures compliance with:
- FDA safety requirements
- Performance and labeling standards
- Risk management expectations
Types of US FDA 510(k) Submissions:
- Traditional 510(k): The standard submission used when there are no special circumstances or modifications.
- Abbreviated 510(k): Leverages FDA guidance documents and consensus standards for compliance.
- Special 510(k): For modifications made to existing devices without changing the intended use.
Why US FDA 510(k) is Important for Medical Devices?
For Omani manufacturers, US FDA 510(k) clearance offers multiple benefits:
- Legal entry into the U.S. medical device market
- Compliance with stringent FDA safety and performance standards
- Faster regulatory approval compared to PMA
- Enhanced global credibility and investor confidence
Whether you manufacture IVD test kits, surgical instruments, or diagnostic devices, US FDA 510(k) clearance is a key step toward international expansion.
How Operon Strategist Supports US FDA 510(k) Submission:
At Operon Strategist, we act as your dedicated 510k consultant to help streamline every step of your USFDA 510(k) process:
- End-to-end FDA dossier preparation and review
- Guidance on Traditional, Abbreviated, and Special 510(k) pathways
- Support with Establishment Registration and Device Listing
- Consulting for Premarket Approval (PMA) and De Novo submissions for innovative devices
- Expertise in FDA 510(k) assays and IVD regulatory documentation
We ensure that your submission meets all FDA requirements, reducing the chances of delays or rejections.
Need More Clarity on the Process of US FDA 510(k)?
Why Choose Operon Strategist for US FDA 510(k) Clearance in Oman?
Operon Strategist is a trusted US FDA 510(k) consultant in Oman, supporting medical device and IVD manufacturers with accurate, compliant, and timely FDA submissions. Our regulatory experts understand both US FDA expectations and the practical challenges faced by Omani manufacturers, ensuring a smooth and efficient US FDA 510(k) clearance journey.
Beyond US FDA 510(k) consulting, we offer integrated regulatory and compliance services, including Medical Device Design and Development Consulting, ISO 13485 QMS Implementation, CAPA Management Consulting, and Post-Market Surveillance Services to support your product throughout its entire lifecycle. We also assist with CE Marking under EU MDR, FDA Audit and Inspection Support, and Turnkey Medical Device Regulatory Consulting, making us a single-point solution from concept to commercialization.
With Operon Strategist, you gain a long-term regulatory partner who helps you achieve US FDA 510(k) approval, maintain ongoing compliance, and expand confidently into global markets.
FAQs
What is US FDA 510(k) clearance?
FDA 510(k) clearance is a regulatory requirement by the U.S. Food and Drug Administration that allows manufacturers to legally market their Class I (non-exempt) and Class II medical devices in the United States. It involves demonstrating that the new device is substantially equivalent to a legally marketed predicate device.
Who needs an FDA 510(k) submission in Oman?
Any Omani medical device manufacturer planning to sell Class II or selected Class I devices, including In Vitro Diagnostic (IVD) products like reagents and test kits, in the U.S. must submit a 510(k) application and receive clearance.
What types of 510(k) submissions are there?
There are three types of FDA 510(k) submissions:
Traditional 510(k): Standard pathway for most devices.
Abbreviated 510(k): Uses recognized standards and FDA guidance.
Special 510(k): For changes to a device already cleared, with the same intended use.
Why is FDA 510(k) clearance important for Omani manufacturers?
FDA 510(k) clearance enables legal marketing in the U.S., ensures compliance with FDA regulations, and enhances your brand's credibility in the global market. It also helps in faster market entry and competitive positioning.
Is FDA 510(k) different from CE marking?
Yes. FDA 510(k) is specific to the U.S. market, while CE marking applies to medical devices sold in the European Union. Both have different regulatory frameworks, though some documentation may overlap.