

EU MDR Implementation: An Overview
The European Union Medical Device Regulation (EU MDR) indeed represents a substantial shift in the regulatory landscape for medical device manufacturers, importers, distributors, and authorized representatives within the European market. This regulation, implemented on May 26th, 2021 (delayed from the original date of May 26th, 2020), is a significant overhaul of the existing regulatory framework, aiming to enhance patient safety and strengthen the approval process for medical devices.
The transition from the Medical Device Directive (MDD) to the EU MDR necessitates a comprehensive understanding and adaptation by all stakeholders involved in the medical device industry. Some key points to consider include:
Looking for an EU MDR consultant?
Let’s have a word about your project
Changes Introduced by EU MDR:
Regulation vs. Directive: The shift from a directive to a regulation implies that the EU MDR carries legal binding force in its entirety across all EU member states without the need for transposition into national law, ensuring more uniform application and interpretation.
Stringent Requirements: The EU MDR introduces more stringent requirements regarding device classification, conformity assessment procedures, clinical evidence, post-market surveillance, and transparency.
Economic Operator Roles: Manufacturers, importers, distributors, and authorized representatives all have distinct responsibilities under the EU MDR. Compliance requirements vary based on these roles.
Key Focus Areas: The implementation process involves several critical areas such as device classification, creating technical files, risk management in accordance with ISO 14971, Unique Device Identification (UDI) system, post-market surveillance (PMS), post-market clinical follow-up (PMCF), clinical evaluation, labeling, and participation in EUDAMED.
Step-By-Step Guide to EU MDR Implementation:
Below you will find a step-by-step EU MDR IMPLEMENTATION guide with regards to the new medical device regulation (MDR EU2017/745). Our guide is simple to understand and will allow you to save time and money when implementing the new regulation.
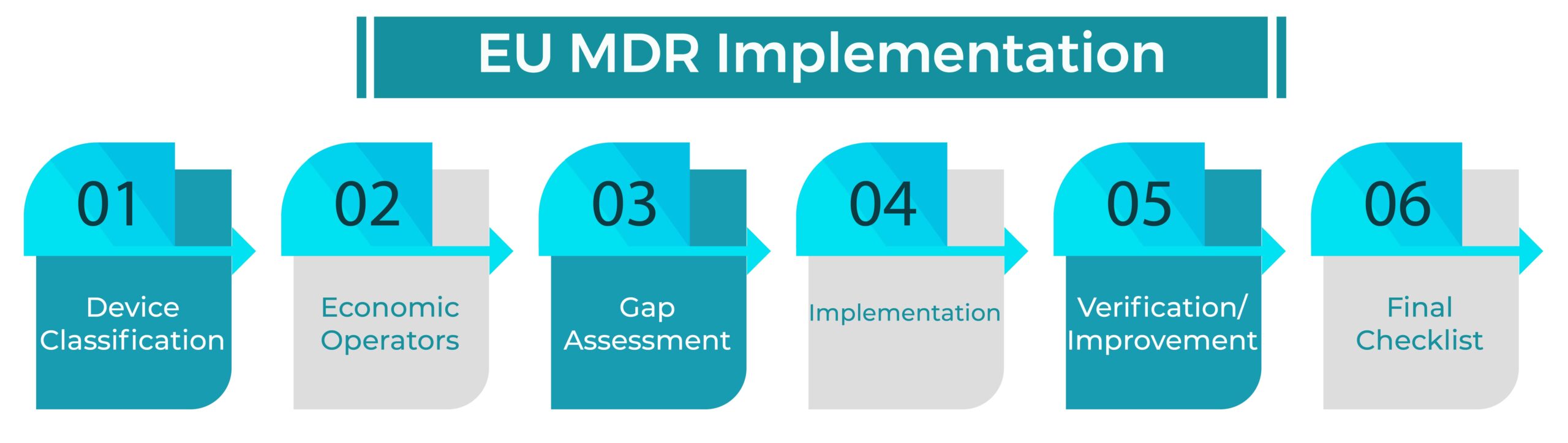
Step 1: DEVICE CLASSIFICATION
In a first step you should check if the new MDR rules have any impact on your existing or future products classifications.
Step 2: ECONOMIC OPERATORS
Depending on your business you may have one or multiple economic operator responsibilities, which is imperative to know before you start with the gap assessment. Economic operators are Manufacturers, Importers, Distributors or Authorized Representative. Each of them has different requirements to fulfill per the EU MDR Implementation.
Step 3: GAP ASSESSMENT
- Reduce not required information by going through the chapters and annexes and eliminate all not required information.
- Define keywords which are not applicable to you. In our MDR tool you have the opportunity to search for these keywords.
- Go through all open requirements step by step and define if requirements are relevant for your business or not.
Step 4: IMPLEMENTATION:
Prior to starting the implementation phase, you should put a plan in place. The steps below will guide you through the main topics.
- Safety and performance checklist
- Technical File
- Risk management according to ISO 14971
- UDI System
- Post-Market Surveillance (PMS)
- Post-Market Clinical Follow-Up (PMCF)
- Clinical Evaluation
- Labelling
- EUDAMED (European database on medical devices)
- Common Specifications
Step 5: VERIFICATION / IMPROVEMENT
Performing internal audits and a final mock audit to ensure the key requirements have been implemented.
Step 6: FINAL CHECKLIST:
Make a final “written” checklist, where you can show the evidence for each requirement.
Empower your EU MDR compliance journey with Operon Strategist’s expert consultancy. Contact us today for tailored solutions, seamless implementation, and guidance from experienced EU MDR consultants. Let us navigate the complexities, ensuring your adherence to regulations for a successful market entry.


