

How to Manage ISO 13485 Design Changes in Medical Devices and Stay Compliant?
Changes in medical device design, manufacturing processes, or regulatory requirements are inevitable. Whether your company is a startup or an established manufacturer, effective change control is crucial to ensure compliance with ISO 13485 design changes and FDA 21 CFR Part 820.30(i). Without proper change management, companies risk regulatory violations, safety concerns, and product recalls.
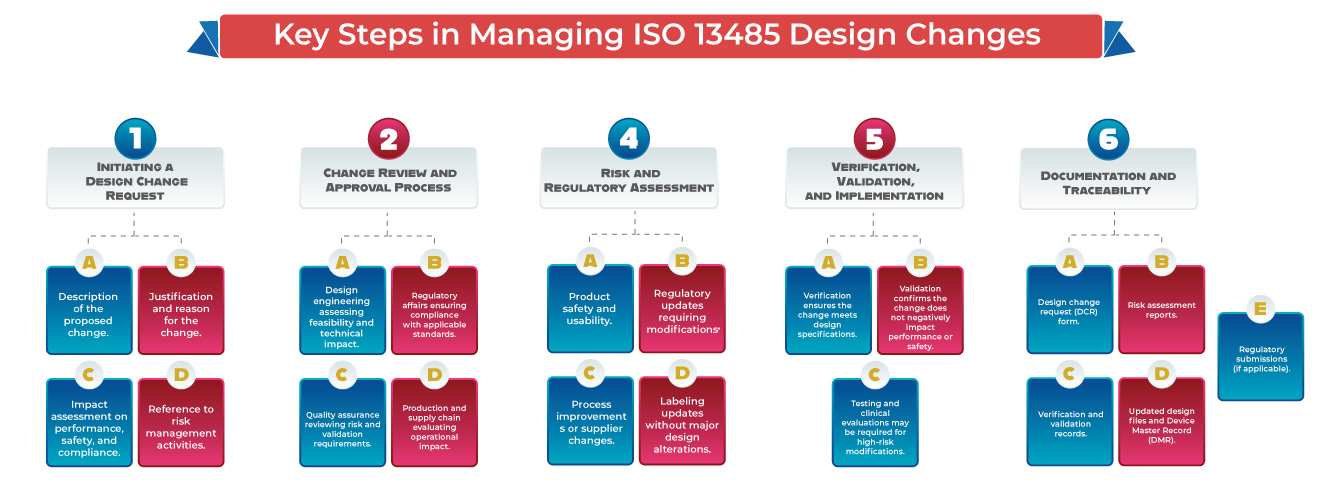
This article outlines the key steps and best practices for managing ISO 13485 design changes while meeting international regulatory requirements.
Looking For a Medical Device Regulatory Consultant?
Let’s have a word about your next project
Why Change Control Matters for Medical Devices?
- Medical devices must meet strict quality and safety standards. Any design or process change, if not properly managed, can impact product performance, patient safety, and regulatory compliance. A well-structured change control system ensures:
- Formal documentation and approval of all requested changes.
- Risk assessment and validation before implementation.
- Compliance with global regulatory standards (ISO 13485 design changes, USFDA QSR, EU MDR, etc.).
- Traceability and audit readiness for regulatory inspections.
Regulatory Requirements for ISO 13485 Design Changes
ISO 13485:2016 (Clause 7.3.9) – Control of Design and Development Changes
According to ISO 13485 design changes requirements, manufacturers must establish procedures to control design changes. The standard requires:
- Identification and documentation of changes.
- Review of potential impact on safety, usability, and regulatory compliance.
- Verification and validation before implementation.
- Formal approval and traceability of changes.
FDA 21 CFR Part 820.30(i) – Design Changes
Under FDA regulations, medical device manufacturers must:
- Identify, document, and assess changes.
- Conduct risk-based validation or verification.
- Ensure changes are reviewed and approved before implementation.
- Maintain records of all ISO 13485 design changes modifications.
Failure to comply with these requirements may result in FDA warning letters, recalls, or regulatory penalties.
How Operon Strategist Supports Your ISO 13485 Design Changes Process
Operon Strategist helps medical device manufacturers manage design changes while ensuring compliance with ISO 13485:2016 and FDA 21 CFR Part 820.30. Our experts assist with:
- Change Control Implementation – Establishing a structured process for reviewing, approving, and documenting design modifications.
- Regulatory Compliance – Ensuring changes meet global regulatory standards.
- Risk Assessment – Identifying potential risks and mitigation strategies.
- Validation & Verification – Supporting testing and documentation of design updates.
Our Additional Services
We also provide:
- ISO 13485 QMS Implementation
- FDA 510(k) Submission
- CE Marking for Medical Devices
- Medical Device Risk Management
- Process Validation & Cleanroom Setup
With Operon Strategist, you get expert guidance to streamline approvals, ensure compliance, and maintain product quality.

