
US FDA 21 CFR 820.30 Design Control Requirements
What is US FDA 21 CFR 820.30 Design Controls For Medical Devices?
FDA 21 CFR Part 820.30 design control medical device requirements are the most important stage in advancing a medical device since a defective plan may prompt it to be inadequate or dangerous (not affirmed or cleared by the administrative organization). At the design stage, an outline design control process should be started and actualized as a feature of the Quality System Requirement. Generally, outline design controls are straightforward and logical steps to ensure that what you develop is what you meant to create and that the last item lives up to your client’s needs and desires.

We have experience with each constituent part and the GMP regulations that together form the basis for their development and manufacture: Drug-Device Combination Products (FDA 21 CFR part 820) and (21 CFR Part 4).
Read Ultimate Guide to Design Control for Medical Device Companies
What is the Process for Medical Device Design Controls 21 CFR 820.30?
Build up and maintain a plan that describes the design and development activities and allocates the individual obligations for each activity. Guarantee you review, update, and approve the plan until the device design is completed, verified, and validated.
Looking for Medical Device Consultant?
What are FDA Design Controls for Medical Devices?
The design control process conforms to a set of practices and procedures that aid medical product developers in the following ways:
- Manage quality.
- Ensure each product meets all requirements.
- Prevent potential issues or recalls in the future.
FDA Design Control Guidance for Medical Devices
The FDA is in charge of regulating design controls for medical devices under 21 CFR 820.30. Manufacturers of class II or class III medical equipment must install them (and some class I devices).
Outside of the United States, ISO 13485:2016 establishes a set of rules that are extremely similar (almost identical, in fact). We’ll concentrate on the FDA rules in this case to keep things as straightforward as possible.
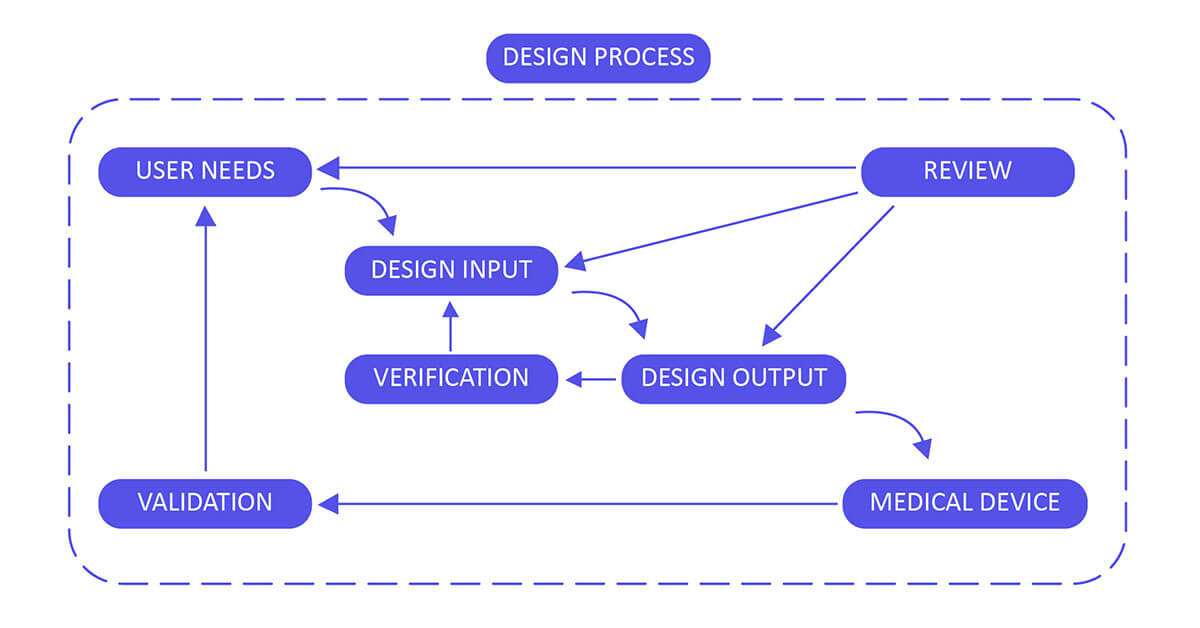
Medical Device Design Controls – 21 CFR 820.30 Process
Design Input :
Utilize performance, safety, business economics, outputs of risk management and regulatory requirements as a basis to plan the device with the goal that its motivation and the proposed utilize are clear. The input may also come from surveying your customers (For example, clinicians, nurses, patients).
Design Output :
Design output methods or particulars need to stipulate or refer to the design input document developed by the team and need to identify the critical measures/outputs for the best possible capacity of the device. These incorporate the tests and strategies that may have been produced, adjusted or used to show conformance with the characterized configuration inputs. Examples of design outputs may include:
- The device itself.
- The user manual.
- Specifications A Risk Analysis Study results (For examples, validation and biocompatibility studies, storage).
- Technical Files.
Design Review :
Confirm the design, or identify at an opportune time and right any insufficiencies distinguished at other plan and improvement phases. Two common types of review are hazard analysis, and failure mode and effect analysis.
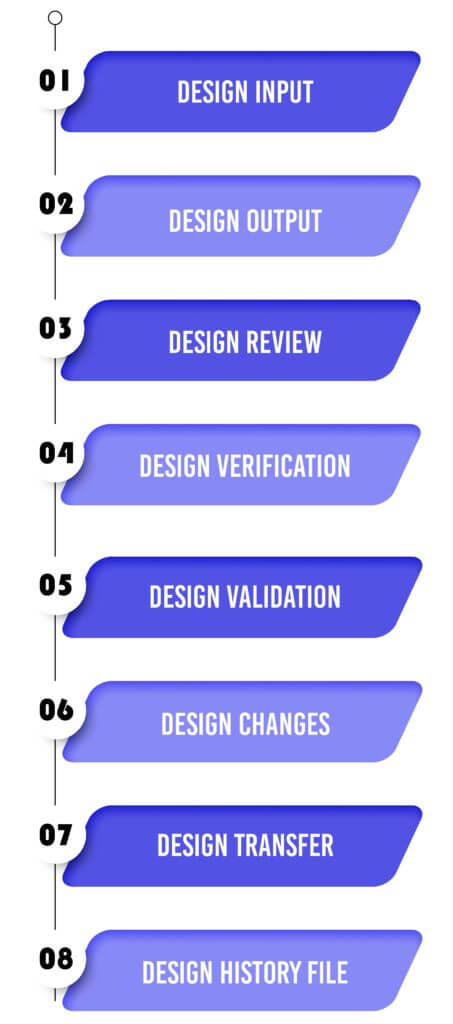
Design Verification :
Confirm the device outline by means of examination and target prove, verify that the design outputs meet the plan inputs. Design verification activities must be arranged and routinely analyzed and the outcomes must be documented.
Design Validation :
Approve the device design plan by means of examination and target prove, affirm that the last outline yield reliably meets the particular planned utilize. Design validation should follow successful design verification. Since outline check is directed while the plan work is being performed, design validation confirms that the medical device meets its intended use. Generally, this is set up through in vitro execution, practical testing, creature testing and additionally in vivo clinical assessments and trials.
Design Changes :
Guarantee that all plan changes are distinguished, documented, approved, verified, reviewed and endorsed before usage.
Design Transfer :
Ensure that the design of the medical device can be correctly translated into production specifications (that is, advancing successfully from product development to manufacturing).
Design History File :
The design history file (DHF) aggregates confirm (that is, the history of the design) that demonstrates that the outline was created as per the outline of design control― specifically, the design and development plan, or the outline change design.
Operon Strategist is the leading medical device regulatory consultant providing consultation for 21 CFR 820.30 design controls Medical Devices with extensive experience and the practical implementation of design controls regulation for developing new design control processes or for making improvements to existing processes. We are a medical device regulatory consultant and assist in CDSCO registration. We assist in design controls as per FDA and ISO 13485:2016 that can be mapped to the process that works best for the organization and the product being developed. If you need any help setting up a design control system or wish to modify an existing system in order to align with ISO 13485 or FDA design controls. Contact us now.
Get FDA-Compliant Design Controls for Your Medical Device
FAQs:
What is Design Control for Medical Device?
Design Control labels the implementation of a formalistic technique to the ways of product development activities. It is often necessary by regulation for the appliance of such practice when designing and developing products within regulated industries e.g. medical devices. The Food and Drug Administration FDA has stated that medical device manufacturers that want to marketized certain classifications of medical devices in the USA follow design control requirements (21 CFR 820.30). Few firms regard Risk Management and Design Control as they are linked together but they follow separate procedures, not actualizing that the relation between the user needs, design inputs, dangers and dangerous circumstances. Design control conditions that when the suppliers or manufacturers get a product to design controls they may discover and maintain the correct documentation to assure the specified design requirements are.
What is the difference between 21 CFR 820 and ISO 13485?
ISO: ISO 13485 is the worldwide acknowledged standard by the International Standards Organization for medical device Quality Management System. The standard states the things required for QMS that helps companies execute and exhibit the abilities to provide high-quality medical devices that reach up to customers and regulatory requirements. ISO 13485 can be used by any company involved in the transfer of medical devices, providing, support phases. Furthermore, it can also be used by external or internal auditors to support the inclusive audit process.
21 CFR Part 820: FDA 21 CFR Part 820: covers up the process used in & the facilities and controls used for the design, manufacture, packaging, labelling, storage, installation, & servicing of medical devices. Manufacturers are inspected by the US FDA as per Part 820 & only compliance with the requirements is assessed. FDA 21 CFR known as the Quality System Regulation QSR acknowledges Current Good Manufacturing Practices CGMP regulations that govern the methods used in and the services, and controls used for, the design, manufacturing, packaging, labelling, storage, installation and servicing for all finished devices willful for human use. These requirements are necessary to ensure that medical devices are safe and effective for use. Medical device manufacturers undergo FDA inspections to assure FDA 21 CFR 820 agreement.
What are Examples of Design Controls?
Examples include, diagrams, drawings, specifications and procedures. The outputs from one stage may become inputs to the next stage. The total finished design output consists of the device, its packaging and labeling, and the device master record.
Are Design Controls Part of QMS?
Design Controls is one process that should be integrated into the quality management system (QMS). Your QMS should also have procedures in place to manage documents, records, and suppliers, so Design Controls link those pieces together with additional development specific processes.