European CE Mark Medical Device (EU MDR) Consultant
EU MDR for Medical Devices
Operon Strategist, a leading EU MDR consultant, boasts a substantial presence throughout the EU, extending our support to countless medical device manufacturers to ensure CE compliance for Europe.
What Is CE Marking for Medical Devices?
Selling medical devices in the European Union (EU) requires obtaining CE marking for your medical devices. CE stands for Conformité Européenne. CE marking indicates that your medical device complies with applicable EU regulations and enables the commercialization of your products across all EU member states.
As a legal medical device manufacturer, you are responsible for maintaining regulatory compliance and securing CE mark for your medical devices, regardless of whether you outsource any or all components of your manufacturing operation. Manufacturers of in vitro diagnostic (IVD) devices must meet similar requirements for CE IVD certification in Europe.
Recommended blog: IVD CE Marking: Compliance Certification for In Vitro Diagnostic Devices in Europe

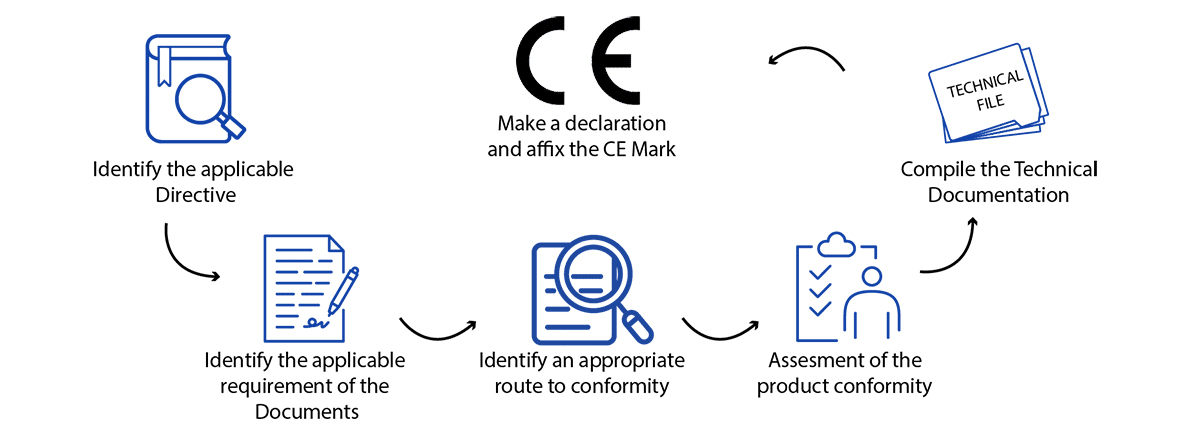
How to Get a CE Mark for a Medical Device?
While “CE” isn’t a quality mark, adherence to the EU Medical Devices Regulation (MDR 2017/745) necessitates meeting precise standards of performance, quality, safety, and efficacy tailored to your product type. Operon Strategist’s comprehensive guide outlines the current European CE approval process for medical devices. Nonetheless, the fundamental procedure for obtaining a CE mark for medical devices typically involves the following steps:
- Determine if your product aligns with the definition of a medical device as outlined in the EU MDR.
- Determine the medical device classification in Europe for your medical devices.
- Implement a Quality Management System (QMS) if it applies to your device. Many companies utilize ISO 13485 to fulfill these requirements.
- Prepare a CE Marking Technical File or a Design Dossier.
- Compile a Clinical Evaluation Report (CER) following the guidelines of MEDDEV 2.7/1 rev4 and the MDR.
- Select and designate a European Authorized Representative (EC REP) to act on your behalf within the EU if you lack a physical presence in Europe.
- Subject your QMS and Technical File/Design Dossier to an audit by a Notified Body, unless your device falls under Class I, is non-sterile, and lacks a measuring function.
- Obtain CE certificate for medical devices and ISO 13485 certificates from your designated Notified Body.
- Prepare a EU Declaration of Conformity (DoC) confirming that your device complies with the regulations outlined in the MDR.
If you need more clarity on obtaining CE certification for your medical devices, contact us on WhatsApp or call 918767980322
Declaration of Conformity
After ensuring that the safety and performance criteria have been satisfied and the requisite technical documentation is ready, the manufacturer is obligated to date, sign, and maintain a declaration of conformity. This declaration serves as proof that the product adheres to the standards outlined in the executive order.
Recommended blog: EU Declaration of Conformity for Medical Devices
Quality Management Systems - Requirements for Regulatory Purposes.
- ISO 13485:2016 – The ISO 13485:2016 standard lays out the rules for how to manage the quality of medical devices. Most medical device makers follow this standard because it’s a good way to make sure they meet the quality management system (QMS) requirements in the MDR.
- FDA 21 CFR Part 820 – FDA 21 CFR Part 820 sets the quality system rules for medical device manufacturers. This regulation is the current standard for quality management systems for medical devices used in the United States.
European Authorized Representative (EC REP) Services
For medical device manufacturers or importers without a physical presence in the EU, appointing a European Authorized Representative (EAR) is mandatory for market entry. Operon Strategist offers comprehensive EC REP services to ensure regulatory compliance and streamline the EU registration process under MDR (2017/745) and IVDR (2017/746).
As an EAR, Operon Strategist serves as the point of contact between non-EU manufacturers and EU authorities, managing device registration, verifying Declarations of Conformity, and maintaining technical documentation. Our services include regulatory liaison, support in incident reporting, and ensuring compliance through the product lifecycle, adding our details on product labels and regulatory documents. Freyr’s dedicated EC REP services provide the expertise and support essential for seamless EU market access.
Countries That Accept the CE Marking for Medical Devices
The Following Countries Currently Require CE Mark for Medical Devices: Austria (since 1995), Belgium, Bulgaria (since 2007), Czech Republic (since 2004), Cyprus(since 2004), Denmark, Estonia (since 2004), Finland (since 1995), France, Germany, Greece, Hungary (since 2004), Ireland, Italy, Latvia (since 2004), Lithuania (since 2004), Luxembourg, Malta (since 2004), The Netherlands, Poland (since 2004), Portugal, Romania (since 2007), Slovakia (since 2004), Slovenia (since 2004), Spain, Sweden (since 1995), Croatia (since July 1, 2013)
EEA Countries Requiring CE Mark for Medical Devices: Iceland, Liechtenstein
Special Cases:
Switzerland: Despite not being an EU member or EEA signatory, Switzerland has incorporated the Medical Devices Directives into its national legislation, necessitating CE marking for specific medical devices.
Turkey: Although not part of the EU or EEA, Turkey has adopted many of the European CE marking directives. Consequently, CE marking is mandatory for numerous medical devices in Turkey.
Get Your Medical Device CE Marked Fast and Easy With Operon Strategist!
Streamline EU MDR Compliance With Operon Strategist
To simplify the approval process, Operon Strategist, a leading EU MDR consultant, supports manufacturers in the following:
- Assisting with medical device classification
- Verifying applicable standards and testing requirements
- Compiling Technical Files or reviewing existing documentation
- Reviewing marketing materials, labels, and user manuals for compliance and consistency
- Ensuring adherence to Essential Requirements
- Crafting Clinical Evaluation Reports based on provided clinical data
- Implementing, adjusting, and maintaining a quality system (usually ISO 13485) to meet European and international criteria
- Providing European Authorized Representative services
- Conducting risk assessment and management following ISO 14971
- Developing vigilance and post-market surveillance procedures
MDR CE marking Approval process will vary according to the Class of medical device, as per the EU MDR 2017/745, CE marking Approval consist of a few more things such as product quality, technical dossier submission to Notified Body, clinical evaluation, and so on.
Contact us to learn how we can assist you in obtaining CE marking for your medical device in Europe.
Simplify Your CE Mark Journey
Get expert guidance on CE marking for medical devices. From classification to clinical evaluations, technical documentation, and EC REP services, we handle it all. Ensure compliance with EU MDR seamlessly.
Get Your Medical Device CE Marked Effortlessly!
Mail Us:
Call Us Now:
Let's Connect! Your Queries, Our Expertise!
FAQS
1. Check the CE mark’s placement and appearance
2. Verify the identification number of the Notified Body
3. Check the product’s documentation
4. Check the manufacturer’s address
In medical terms, “CE” stands for “Conformité Européene,” which translates to “European Conformity” in French. All medical devices and IVDs, with the exception of custom-made devices and those for clinical investigations, are required to display the CE mark.
Yes, medical devices generally need to be CE marked. The CE marking indicates that a medical device conforms to the essential safety and performance requirements outlined by European Union (EU) regulations. However, there are some exceptions, such as custom-made devices and those intended for clinical investigations, which may not require CE marking.
Typically, under the current framework, CE certificates issued by Notified Bodies remain valid for about three years. However, for certain high-risk devices, this validity period might be reduced to one year. It’s important to note that the continuity of your CE certification relies on upholding your quality system certification.
The duration to acquire a CE mark for a medical device typically spans four to six weeks on average. However, the timeline can vary, influenced by factors such as test outcomes, necessary product adjustments, and the promptness of providing technical documentation. It’s important to note that the specific CE marking process varies for each product.
For Class 1 medical devices, obtaining CE marking can be accomplished through self-declaration according to the MDR. This means that neither Notified Body certification nor approvals from certification bodies are necessary. Class 1 devices are considered to have minimal risk, allowing manufacturers to self-certify them.