

What is Establishment Registration?
Every medical device manufacturer and distributor is required to register their organization with the FDA before selling their devices. Registration must be completed electronically through the FDA Unified Registration and Listing System (FURLS system), and approval from the FDA is necessary for operations to commence.
Annual verification of registration details is mandatory, with a flexible timeframe offered by the FDA—verification can be conducted anytime between October 1st and December 31st each year. The FDA levies registration fees, which are subject to periodic revisions and are a significant aspect of compliance for these establishments.
Looking For US FDA Establishment Registration Consultants?
Let’s have a word about your next project
Medical Device Listing
As part of the medical device listing mandate, organizations must furnish information about the devices they manufacture. This includes providing a premarket submission number, such as 510(k), PMA (Premarket Approval), HDE (Humanitarian Device Exemption), etc., to the FDA if the device necessitates premarket procedures.
Who Needs to Register With the FDA?
As a general rule, if you manufacture all or part of a medical device sold in the US, or perform processing (e.g., sterilization) on the device, you must register.
Except for Initial Importer, FDA also requires registered establishments to list the devices. An initial importer who is responsible for furthering the marketing of a device entering the US from a foreign manufacturer to the final distributor for sale of the device to the ultimate consumer or user must register their establishment but is not required to list the devices.
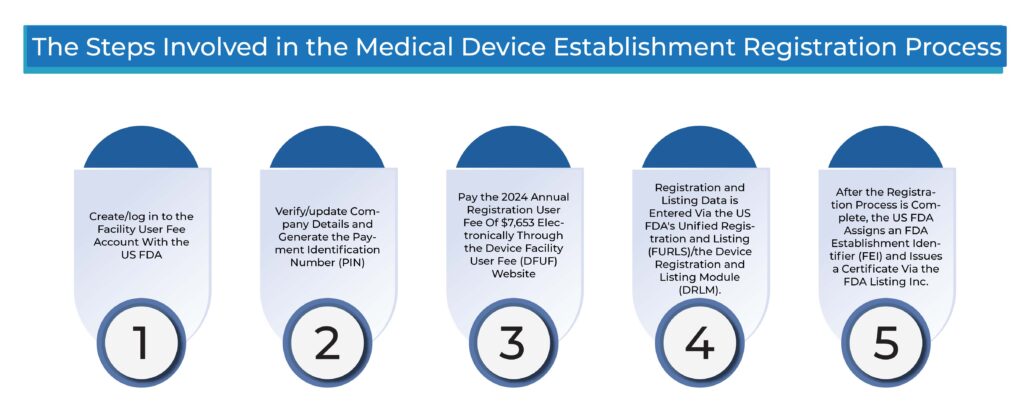
What Are the Steps Involved in the Medical Device Establishment Registration Process?
- Create/log in to the Facility User Fee Account with the US FDA
- Verify/update Company Details and Generate the Payment Identification Number (PIN)
- Pay the 2024 Annual Registration User Fee Of $7,653 Electronically Through the Device Facility User Fee (DFUF) Website.
- Registration and Listing Data is Entered Via the US FDA’s Unified Registration and Listing (FURLS)/the Device Registration and Listing Module (DRLM).
- After the Registration Process is Complete, the US FDA Assigns an FDA Establishment Identifier (FEI) and Issues a Certificate Via the FDA Listing Inc.
Also, read about A Guide to FDA eSTAR Submission Template
How Operon Strategist Will Help You?
Partnering with Operon Strategist can significantly streamline the intricate process of US FDA establishment registration for medical devices. Our expertise in developing regulatory strategies, ensuring documentation compliance, navigating quality management systems, facilitating communication with regulatory authorities, and supporting companies through audits and post-market compliance fosters a smoother path to successful registration. By leveraging our specialized guidance, companies can confidently navigate the complex regulatory landscape, ensuring adherence to FDA standards and expediting the approval process for their medical devices.


